The simple input format consists of keywords followed by a ":" followed by a value. The keywords are case sensitive. The values might be modified by options found in parenthesis. For example, the following input performs an optimization of water using density functional theory with the B3LYP exchange-correlation functional:
% B3LYP optimization of water
optimize: yes
method: KS (xc = B3LYP)
basis: 3-21G*
molecule: (angstrom)
O 0.172 0.000 0.000
H 0.745 0.000 0.754
H 0.745 0.000 -0.754
Comments begin with a % and continue to the end of the line. Keyword values, such as method names, method options, or basis set names containing special characters, such as a space, parentheses, or square braket, must be quoted inside a pair of double quotes:
% DZP CCSD(T)_R12 energy of H2
optimize: no
method: "CCSD(T)_R12" (f12="stg-3g[1.0]")
basis: "DZP (Dunning)"
auxbasis: cc-pV6Z (uc)
molecule:
H 0.000 0.000 0.700
H 0.000 0.000 -0.700
The accepted keywords are:
moleculeGives the atoms types and coordinates. The following options can be used
bohr- The coordinates are given in Bohr.
angstrom- The coordinates are given in Angstroms (the default).
charge- This option can be given after an "element x y z" quadruple. This will override the charge on the atom. For example,
(charge = 0)can be given for the ghost atoms in a counterpoise correction calculation.
multiplicityGives the multiplicity of the molecule. The default is
1.chargeSpecificies the charge on the molecule. The default is
0.optimizeIf
yes, then an optimization will be performed. The default isno. The following options can be given:- Choice of coordinates
cartesian- Use Cartesian coordinates.
internal- Use internal coordinates.
redundant- Use redundant internal coordinates.
convergence- A floating-point number that specifies the convergence thresholds. The default is 1e-4.
- See also
- sc::Convergence
gradient If yes, then a gradient calculation will be performed. The default is no.
frequencies If yes, then the frequencies will be obtained. The default is no. The following options can be given:
accuracy- A floating-point number that specifies the desired relative accuracy of the computed frequencies. The default is 1e-5.
precise_findif Set to yes if need to compute numerical gradient/frequencies computation to high precision. The default is no.
accuracy Specifies an estimate of the accuracy with which the wave function should be computed. The value should be given as a real number, which specifies the target accuracy for the energy. The default is 1e-6 (same as the default for value_accuracy keyword of Function class). The accuracy is not guaranteed to be achieved and thus the user is advised caution.
method Specifices the method. There is no default and the possible values are:
HF- Hartree-Fock. Unrestricted HF is used if
multiplicity> 1 RHF- Restricted Hartree-Fock.
UHF- Unrestricted Hartree-Fock.
KS- Kohn-Sham. Unrestricted KS is used if
multiplicity> 1 RKS- Restricted Kohn-Sham.
UKS- Unrestricted Kohn-Sham.
MP2- Second-order Møller-Plesset perturbation theory. Unrestricted MP2 is used if
multiplicity> 1 UMP2- Unrestricted MP2. No gradient, optimization, of frequencies are possible.
RMP2- Restricted MP2. No gradient, optimization, of frequencies are possible.
ZAPT2- Z-averaged second-order perturbation theory. Only available for
multiplicity> 1. No gradient, optimization, or frequencies are possible. MP2- Second-order Møller-Plesset perturbation theory. Unrestricted MP2 is used if
multiplicity> 1 MP2-R12- The explicitly-correlated MP2 method (
RMP2-R12andUMP2-R12, the R12 equivalents ofRMP2andUMP2, are also allowed). An auxiliary basis should be specified for this method (seeauxbasiskeyword). No gradient, optimization, or frequencies are possible. CCSD(2)_R12- The explicitly-correlated CCSD method (
RCCSD(2)_R12andUCCSD(2)_R12are also allowed). An auxiliary basis should be specified for this method (seeauxbasiskeyword). No gradient, optimization, or frequencies are possible. CCSD(T)_R12- The explicitly-correlated CCSD method (
RCCSD(T)_R12andUCCSD(T)_R12are also allowed) An auxiliary basis should be specified for this method (seeauxbasiskeyword). No gradient, optimization, or frequencies are possible. CC3(2)_R12- The explicitly-correlated (ground-state) CC3 method (
RCC3(2)_R12andUCC3(2)_R12are also allowed). An auxiliary basis should be specified for this method (seeauxbasiskeyword). No gradient, optimization, or frequencies are possible.
The following options are valid with the KS, RKS, and UKS methods:
grid- Specifies the grid to be used for numerical integrations. The following values can be given:
xcoarsecoarsemediumfinexfineultrafine
xc- Specifies the exchange-correlation functional. There is no default. See the table in the StdDenFunctional class documentation for the possible values.
The following options are valid with R12 methods:
f12Specifies the correlation factor used to construct the geminals. The default value is
stg-6g[1.3](geminal exponent of 1.5 is a compromise value that seems to work well with most orbital basis sets; if the specialized R12-optimized basis sets of Peterson are used, the respecive recommended values will be used automatically). The following values can be given:stg-Ng[Y]- Use a Slater-type geminal,
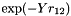 , where Y is the orbital exponent (typical values between 0.5 and 2.0), expanded in terms of N Gaussian-type geminals (N must be greater or equal to 1). The use of this correlation factor requires that the Libint2 library was used to configure and compile MPQC.
, where Y is the orbital exponent (typical values between 0.5 and 2.0), expanded in terms of N Gaussian-type geminals (N must be greater or equal to 1). The use of this correlation factor requires that the Libint2 library was used to configure and compile MPQC. r12- Use the linear correlation factor, which was used in the original R12 theory. The linear factor is less effective than the Slater-type geminal and should only be used by experts. The use of this correlation factor requires that the Libint1 library was used to configure and compile MPQC.
none- R12-dependent terms will not be used. This is only useful for comparing with the standard methods or accessing certain features (e.g. DF-MP2) not available with the conventional implementations.
appSpecifies the R12 approximation. The default is
app=Cand should be fine for most non-expert uses. The following values can be given:A'- Use approximation A'.
A''- Use approximation A'' (unpublished method by Valeev).
B- Use approximation B.
C- Use approximation C. This is the default.
ansatzSpecifies the R12 ansatz used to generate the geminal functions. The following values can be given:
diag- Use the diagonal orbital-invariant ansatz with coefficients fixed by the first-order cusp-conditions (due to Ten-no). This is the default and recommended for most non-experts.
ijij- Same meaning as
diag. ijkl- Use the original orbital-invariant ansatz of Klopper.
ijpq- Use pq ansatz.
ri- Specifies the manner in which the resolution-of-the-identity is used. The default is
ri=cabs+and should be used by all non-experts. The following values can be given:abs- Use the Auxiliary Basis Set method of Klopper and Samson.
abs+- Same as
absbut use the union of the orbital and the given auxiliary basis as the actual auxiliary basis set used. cabs- Use the Complementary Auxiliary Basis Set method of Valeev.
cabs+- Same as
cabsbut use the union of the orbital and the given auxiliary basis as the actual auxiliary basis set used. This is the default.
basis Specifies the basis set. There is no default. See the table in the GaussianBasisSet class documentation for the available basis sets. The following options can be given:
uc- Uncontract the basis.
puream- Force the use of solid harmonics for all shells (see
pureamkeyword of GaussianBasisSet). This will be set by default for Psi-based wave functions. split- Split generally-contracted shells (see SplitBasisSet). May be necessary for certain Integral factory types. Will be set automatically when needed.
auxbasis Specifies the auxiliary basis set for R12 methods. There is no default. See the table in the GaussianBasisSet class documentation for the available basis sets. Refer to the documentation for keyword basis for the list of options that can be given.
dfbasis Specifies the density-fitting basis set. There is no default. See the table in the GaussianBasisSet class documentation for the available basis sets. Refer to the documentation for keyword basis for the list of options that can be given.
symmetry Specifices the Schoenflies symbol of the point group of the molecule. The default is auto, which will cause the program to find the highest order Abelian subgroup of the molecule.
docc Gives the number of doubly occupied orbitals in each each irreducible representation in a parenthesized list. The symmetry must be specified and not be auto. The method must be spin-restricted.
socc Gives the number of single occupied orbitals in each each irreducible representation in a parenthesized list. The symmetry must be specified and not be auto. The method must be spin-restricted.
alpha Gives the number of alpha occupied orbitals in each each irreducible representation in a parenthesized list. The symmetry must be specified and not be auto. The method must be spin-unrestricted.
beta Gives the number of beta occupied orbitals in each each irreducible representation in a parenthesized list. The symmetry must be specified and not be auto. The method must be spin-unrestricted.
frozen_docc Gives the number of frozen core orbitals. Can be either a single integer (e.g. frozen_docc=0 will not freeze core orbitals) or a parenthesized list giving the frozen core orbitals in each irreducible representation. In the latter case the symmetry must be given and not be auto. The default is to automatically determine the frozen core orbitals.
frozen_uocc Gives the number of frozen virtual orbitals. Can be either a single integer or a parenthesized list giving the frozen virtual orbitals in each irreducible representation. In the latter case the symmetry must be given and not be auto.
scf Specifies the parameters of the self-consistent field procedure used in all HF and DFT methods. The following options can be used
maxiter- The maximum number of iterations. The default is 40 (same as the default for
maxiterkeyword of SCF class).
lindep Specifies the tolerance used to detect linearly dependent basis functions. All eigevectors of the overlap matrix with eigenvalues smaller than the maximum eigenvalue times this tolerance are eliminated using the symmetric orthogonalization. The default is 1e-8 (same as the default for lindep_tol keyword of Wavefunction class).
restart Set to yes to restart an optimization. The default is no.
checkpoint Set to no to not save checkpoint files during an optimization. The default is yes.
tmpstore Specifies how the large temporary data will be held. The default is disk, i.e. to write such files to disk. Setting this keyword to mem will force the algorithm to hold such data in memory.
tmpdir Gives the directory to which large temporary files will be written. The default is to write such files into the current working directory, i.e. ./ .
memory Gives a hint for the amount of memory in bytes that can be used. This is typically a lower bound, more memory will be used in practice and the exact amount cannot be precisely controlled. The format is a fixed or floating point number optionally followed (without spaces) by one of the following suffixes: KB, MB, GB, KIB, MIB, or GIB.
debug Specifies the amount of information printed out (this should help troubleshooting). Nonnegative integer values are accepted. The higher the value the more output will be produced. The default is 0.