MPQC is an object-oriented program that directly allows the user to specify objects that MPQC then manipulates to obtain energies, properties, etc. This makes the input very flexible, but very complex. However, most calculations should be quite similar to the one of the examples given later in this chapter. The best way to get started is to use one of the example input files and modify it to meet your needs.
The object-oriented input format is described in the following sections:
- Overview of the Object-Oriented Input
- A Walk-Through of an Object-Oriented Input File
- Sample Object-Oriented Input Files
Overview of the Object-Oriented Input
MPQC starts off by creating a ParsedKeyVal object that parses the input file specified on the command line. The format of the input file is documented in the KeyVal documentation(see The KeyVal Library). It is basically a free format input that associates keywords and logical groupings of keywords with values. The values can be scalars, arrays, or objects.
The keywords recognized by MPQC begin with the mpqc prefix. That is, they must be nested between an mpqc:( and a ). Alternately, each keyword can be individually prefixed by mpqc:. The primary keywords are given below. Some of the keywords specify objects, in which case the object will require more ParsedKeyVal input. These objects are created from the input by using their ParsedKeyVal constructors. These constructors are documented with the source code documentation for the class.
moleThis is the most important keyword for MPQC. It specifies the MolecularEnergy object. This is an object that knows how to compute the energy of a molecule. The specializations of MolecularEnergy that are most commonly used are CLKS, HSOSKS, UKS, CLHF, HSOSHF, UHF, and MBPT2.
optThis keyword must be specified for optimizations. It specifies an Optimize object. Usually, QNewtonOpt is best for finding minima and EFCOpt is best for transition states.
freqThis keyword must be specified to compute frequencies. It specifies a MolecularFrequencies object.
threadThis specifies an object of type ThreadGrp that can be used to advantage on shared-memory multiprocessor machines for certain types of calculations. This keyword can be overridden by giving the ThreadGrp in the environment or command line. See the section on running MPQC for more information.
integralsThis specifies an object of type Integral that will be used as the default integral evaluator. If MP2-R12 is used, then this should be set to use IntegralCints with a line like
integrals<IntegralCints>: ().checkpointThe value of this keyword is boolean.
<ul> <li><tt>true</tt> and optimization is to be performed <br> <tt>opt</tt> object will be checkpointed after each iteration. The checkpoint file suffix is ".ckpt". <li><tt>true</tt> and optimization is not performed <br> <tt>mole</tt> object will be checkpointed at intermediate points. The manner in which <tt>mole</tt> will be checkpointed depends on its particular type. The checkpoint file suffix is usually ".wfn", however in general it will depend on the particular specialization of <tt>MolecularEnergy</tt>. </ul> The default is to not checkpoint. </dd>checkpoint_freqThis specifies how often to checkpoint certain MolecularEnergy specializations which compute iteratively. Currently, mole objects of SCF type can use this keyword. The default is 1, which means to checkpoint after every iteration.
savestateThe value of this keyword is boolean. If true, then the states of the Optimize and MolecularEnergy objects will be saved after the calculation completes. The output file suffixes are ".ckpt" and ".wfn", respectively. The default is to save state.
restartThe value of this keyword is boolean. If true, mpqc will attempt to restart the calculation. If the checkpoint file is not found, the calculation will continue as if the value were false. The default is true.
restart_fileThis gives the name of a file from which restart information is read. If the file name ends with ".wfn" then MPQC will try to restore a
MolecularEnergyobject from it and query for theoptobject in the input file. If the file name ends with ".ckpt" MPQC will try to restore anOptimizeobject from this file. The default file name is formed by appending ".ckpt" to the input file name with the extension removed.do_energyThe value of this keyword is boolean. If true a single point energy calculation will be done for the MolecularEnergy object given with the mole keyword. The default is true.
do_gradientThe value of this keyword is boolean. If true a single point gradient calculation will be done for the MolecularEnergy object given with the mole keyword. The default is false.
do_ccaThe value of this keywork is boolean. If true the cca embedded framework will be initialized. The default is false.
cca_pathThe value of this keyword is a string that provides a colon-separated list of directories in which CCA component libraries may be found.
cca_loadThe value of this keyword is a string that provides a colon-separated list of sidl class names for CCA components which will be instantiated from the libraries found in the path given by
cca_path.optimizeThe value of this keyword is boolean. If true and the opt keyword was set to a valid value, then an optimization will be performed. The default is true.
write_pdbThe value of this keyword is boolean. If true a PDB file with the molecular coordinates will be written.
filenameThe value of this keyword is a string that gives a name from which checkpoint and other filenames are constructed. The default is the basename of the input file.
print_timingsIf this is true, timing information is printed at the end of the run. The default is true.
There are also some utility keywords that tell mpqc some technical details about how to do the calculation:
debug- This optional keyword gives a Debugger object which can be used to help find the problem if MPQC encounters a catastrophic error.
matrixkit- This optional keyword gives a SCMatrixKit specialization which is used to produce matrices of the desired type. The default is a ReplSCMatrixKit which replicates matrices on all of the nodes. Other choices are not thoroughly tested.
A Walk-Through of an Object-Oriented Input File
This example input does a Hartree-Fock calculation on water. Following is the entire input, followed by a breakdown with descriptions.
% This input does a Hartree-Fock calculation on water.
molecule<Molecule>: (
symmetry = C2V
unit = angstrom
{ atoms geometry } = {
O [ 0.00000000 0.00000000 0.37000000 ]
H [ 0.78000000 0.00000000 -0.18000000 ]
H [ -0.78000000 0.00000000 -0.18000000 ]
}
)
basis<GaussianBasisSet>: (
name = "STO-3G"
molecule = $:molecule
)
mpqc: (
mole<CLHF>: (
molecule = $:molecule
basis = $:basis
)
)
We start with a descriptive comment. Comments begin with a %. Everything from the % to the end of the line is ignored.
% This input does a Hartree-Fock calculation on water.
Now lets set up a Molecule object. The name of the object comes first, it is molecule. Then, in angle brackets, comes the type of the molecule, which is the class Molecule. The keyword and class name are followed by a : and then several pieces of input grouped between a pair of matching parentheses. These parentheses contain the information that will be given to Molecule KeyVal constructor.
molecule<Molecule>: (
The point group of the molecule is needed. This is done by assigning symmetry to a case insensitive Schoenflies symbol that is used to initialize a PointGroup object. An Abelian point group should be used.
symmetry = C2V
The default unit for the Cartesian coordinates is Bohr. You can specify other units by assigned unit to a string that will be used to initialize a Units object.
unit = angstrom
Finally, the atoms and coordinates are given. This can be given in the shorthand table syntax shown below. The headings of the table are the keywords between the first pair of brackets. These are followed by an = and another pair of brackets that contain the data. The first datum is assigned to the first element of the array that corresponds to the first heading, atom. The second datum is assigned to the first element of the array associated with the second heading, geometry, and so on. Here the second datum is actually a vector: the x, y and z coordinates of the first atom.
{ atoms geometry } = {
O [ 0.00000000 0.00000000 0.37000000 ]
H [ 0.78000000 0.00000000 -0.18000000 ]
H [ -0.78000000 0.00000000 -0.18000000 ]
}
)
Next, a basis set object is given.
basis<GaussianBasisSet>: ( name = "STO-3G" molecule = $:molecule )
Now we will give the main body of input. All the subsequent keywords will be grouped in the mpqc section of the input (that is, each keyword will be prefixed with mpqc:).
mpqc: (
Next we give the mole keyword which provides a specialization of the MolecularEnergy class. In this case we will do a closed-shell Hartree-Fock calculation. That is done with an object of type CLHF. The keywords that CLHF accepts are given with the documentation for the CLHF class, usually in the description of the const RefKeyVal& constructor for the class. Also with the CLHF documentation is a list of parent classes. Each of the parent classes may also have input. This input is included with the rest of the input for the child class.
mole<CLHF>: (
The next line specifies the molecule to be used. There are two things to note, first that this is actually a reference to complete molecule specification elsewhere in the input file. The $ indicates that this is a reference and the keyword following the $ is the actual location of the molecule. The : in front of the keyword means that the keyword is not relative to the current location in the input, but rather relative to the root of the tree of keywords. Thus, this line grabs the molecule that was specified above. The molecule object could have been placed here, but frequently it is necessary that several objects refer to the exact same object and this can only be done using references.
The second point is that if you look at the documentation for CLHF, you will see that it doesn't read molecule keyword. However, if you follow its parent classes up to MolecularEnergy, you'll find that molecule is indeed read.
molecule = $:molecule
Just as we gave molecule, specify the basis set with the basis keyword as follows:
basis = $:basis
Now we close off the parentheses we opened above and we are finished.
) )
Sample Object-Oriented Input Files
The easiest way to get started with mpqc is to start with one of sample inputs that most nearly matches your problem. The src/bin/mpqc/samples contains all of the sample inputs below:
- Hartree-Fock Energy
- MP2 Energy
- MP2-R12 energy
- Hartree-Fock Optimization
- Optimization with a Computed Guess Hessian
- Optimization Using Newton's Method
- Hartree-Fock Frequencies
- Giving Coordinates and a Guess Hessian
- Optimization with a Hydrogen Bond
- Fixed Coordinate Optimization
- Transition State Optimization
- Transition State Optimization with a Computed Guess Hessian
- Hartree-Fock energy with intermediate checkpointing
- MP2-R12 energy with intermediate checkpointing
- HF gradient computed from a previously computed HF wave funtion
- MP2 Energy computed using precomputed Hartree-Fock wave function
- CLHF energy using a CCA integrals component
Hartree-Fock Energy
The following input will compute the Hartree-Fock energy of water.
% emacs should use -*- KeyVal -*- mode
% molecule specification
molecule<Molecule>: (
symmetry = C2V
unit = angstrom
{ atoms geometry } = {
O [ 0.00000000 0.00000000 0.37000000 ]
H [ 0.78000000 0.00000000 -0.18000000 ]
H [ -0.78000000 0.00000000 -0.18000000 ]
}
)
% basis set specification
basis<GaussianBasisSet>: (
name = "STO-3G"
molecule = $:molecule
)
mpqc: (
checkpoint = no
savestate = no
% method for computing the molecule's energy
mole<CLHF>: (
molecule = $:molecule
basis = $:basis
memory = 16000000
)
)
MP2 Energy
The following input will compute the MP2 energy of water.
% emacs should use -*- KeyVal -*- mode
% molecule specification
molecule<Molecule>: (
symmetry = C2V
unit = angstrom
{ atoms geometry } = {
O [ 0.00000000 0.00000000 0.37000000 ]
H [ 0.78000000 0.00000000 -0.18000000 ]
H [ -0.78000000 0.00000000 -0.18000000 ]
}
)
% basis set specification
basis<GaussianBasisSet>: (
name = "STO-3G"
molecule = $:molecule
)
mpqc: (
checkpoint = no
savestate = no
% method for computing the molecule's energy
mole<MBPT2>: (
molecule = $:molecule
basis = $:basis
memory = 16000000
% reference wavefunction
reference<CLHF>: (
molecule = $:molecule
basis = $:basis
memory = 16000000
)
)
)
MP2-R12 energy
The following will compute the MP2-R12 energy of water in standard approximation A' (MP2-R12/A').
% emacs should use -*- KeyVal -*- mode
% molecule specification
molecule<Molecule>: (
symmetry = C2V
unit = angstrom
{ atoms geometry } = {
O [ 0.00000000 0.00000000 0.37000000 ]
H [ 0.78000000 0.00000000 -0.18000000 ]
H [ -0.78000000 0.00000000 -0.18000000 ]
}
)
% basis set specification
basis<GaussianBasisSet>: (
name = "cc-pVDZ"
molecule = $:molecule
)
% auxiliary basis set specification
abasis<GaussianBasisSet>: (
name = "aug-cc-pVDZ"
molecule = $:molecule
)
mpqc: (
checkpoint = no
savestate = no
% method for computing the molecule's energy
mole<MBPT2_R12>: (
molecule = $:molecule
basis = $:basis
aux_basis = $:abasis
stdapprox = "A'"
nfzc = 1
memory = 16000000
integrals<IntegralCints>:()
% reference wavefunction
reference<CLHF>: (
molecule = $:molecule
basis = $:basis
memory = 16000000
integrals<IntegralCints>:()
)
)
)
Hartree-Fock Optimization
The following input will optimize the geometry of water using the quasi-Newton method.
% emacs should use -*- KeyVal -*- mode
% molecule specification
molecule<Molecule>: (
symmetry = C2V
unit = angstrom
{ atoms geometry } = {
O [ 0.00000000 0.00000000 0.37000000 ]
H [ 0.78000000 0.00000000 -0.18000000 ]
H [ -0.78000000 0.00000000 -0.18000000 ]
}
)
% basis set specification
basis<GaussianBasisSet>: (
name = "6-31G*"
molecule = $:molecule
)
mpqc: (
checkpoint = no
savestate = no
% molecular coordinates for optimization
coor<SymmMolecularCoor>: (
molecule = $:molecule
generator<IntCoorGen>: (
molecule = $:molecule
)
)
% method for computing the molecule's energy
mole<CLHF>: (
molecule = $:molecule
basis = $:basis
coor = $..:coor
memory = 16000000
)
% optimizer object for the molecular geometry
opt<QNewtonOpt>: (
function = $..:mole
update<BFGSUpdate>: ()
convergence<MolEnergyConvergence>: (
cartesian = yes
energy = $..:..:mole
)
)
)
Optimization with a Computed Guess Hessian
The following input will optimize the geometry of water using the quasi-Newton method. The guess Hessian will be computed at a lower level of theory.
% emacs should use -*- KeyVal -*- mode
% molecule specification
molecule<Molecule>: (
symmetry = C2V
unit = angstrom
{ atoms geometry } = {
O [ 0.00000000 0.00000000 0.37000000 ]
H [ 0.78000000 0.00000000 -0.18000000 ]
H [ -0.78000000 0.00000000 -0.18000000 ]
}
)
% basis set specification
basis<GaussianBasisSet>: (
name = "6-31G*"
molecule = $:molecule
)
mpqc: (
checkpoint = no
savestate = no
% molecular coordinates for optimization
coor<SymmMolecularCoor>: (
molecule = $:molecule
generator<IntCoorGen>: (
molecule = $:molecule
)
)
% method for computing the molecule's energy
mole<CLHF>: (
molecule = $:molecule
basis = $:basis
coor = $..:coor
memory = 16000000
guess_hessian<FinDispMolecularHessian>: (
molecule = $:molecule
only_totally_symmetric = yes
eliminate_cubic_terms = no
checkpoint = no
energy<CLHF>: (
molecule = $:molecule
memory = 16000000
basis<GaussianBasisSet>: (
name = "3-21G"
molecule = $:molecule
)
)
)
)
% optimizer object for the molecular geometry
opt<QNewtonOpt>: (
function = $..:mole
update<BFGSUpdate>: ()
convergence<MolEnergyConvergence>: (
cartesian = yes
energy = $..:..:mole
)
)
)
Optimization Using Newton's Method
The following input will optimize the geometry of water using the Newton's method. The Hessian will be computed at each step in the optimization. However, Hessian recomputation is usually not worth the cost; try using the computed Hessian as a guess Hessian for a quasi-Newton method before resorting to a Newton optimization.
% Emacs should use -*- KeyVal -*- mode
% molecule specification
molecule<Molecule>: (
symmetry = c2v
unit = angstrom
{ atoms geometry } = {
O [ 0.00000000 0.00000000 0.36937294 ]
H [ 0.78397590 0.00000000 -0.18468647 ]
H [ -0.78397590 0.00000000 -0.18468647 ]
}
)
% basis set specification
basis<GaussianBasisSet>: (
name = "3-21G"
molecule = $:molecule
)
mpqc: (
checkpoint = no
savestate = no
restart = no
% molecular coordinates for optimization
coor<SymmMolecularCoor>: (
molecule = $:molecule
generator<IntCoorGen>: (
molecule = $:molecule
)
)
do_energy = no
do_gradient = no
% method for computing the molecule's energy
mole<CLHF>: (
molecule = $:molecule
basis = $:basis
memory = 16000000
coor = $..:coor
guess_wavefunction<CLHF>: (
molecule = $:molecule
total_charge = 0
basis<GaussianBasisSet>: (
molecule = $:molecule
name = "STO-3G"
)
memory = 16000000
)
hessian<FinDispMolecularHessian>: (
only_totally_symmetric = yes
eliminate_cubic_terms = no
checkpoint = no
)
)
optimize = yes
% optimizer object for the molecular geometry
opt<NewtonOpt>: (
print_hessian = yes
max_iterations = 20
function = $..:mole
convergence<MolEnergyConvergence>: (
cartesian = yes
energy = $..:..:mole
)
)
)
Hartree-Fock Frequencies
The following input will compute Hartree-Fock frequencies by finite displacements. A thermodynamic analysis will also be performed. If optimization input is also provided, then the optimization will be run first, then the frequencies.
% emacs should use -*- KeyVal -*- mode
% molecule specification
molecule<Molecule>: (
symmetry = C1
{ atoms geometry } = {
O [ 0.0000000000 0.0000000000 0.8072934188 ]
H [ 1.4325589285 0.0000000000 -0.3941980761 ]
H [ -1.4325589285 0.0000000000 -0.3941980761 ]
}
)
% basis set specification
basis<GaussianBasisSet>: (
name = "STO-3G"
molecule = $:molecule
)
mpqc: (
checkpoint = no
savestate = no
% method for computing the molecule's energy
mole<CLHF>: (
molecule = $:molecule
basis = $:basis
memory = 16000000
)
% vibrational frequency input
freq<MolecularFrequencies>: (
molecule = $:molecule
)
)
Giving Coordinates and a Guess Hessian
The following example shows several features that are really independent. The variable coordinates are explicitly given, rather than generated automatically. This is especially useful when a guess Hessian is to be provided, as it is here. This Hessian, as given by the user, is not complete and the QNewtonOpt object will fill in the missing values using a guess the Hessian provided by the MolecularEnergy object. Also, fixed coordinates are given in this sample input.
% emacs should use -*- KeyVal -*- mode
% molecule specification
molecule<Molecule>: (
symmetry = C1
{ atoms geometry } = {
H [ 0.088 2.006 1.438 ]
O [ 0.123 3.193 0.000 ]
H [ 0.088 2.006 -1.438 ]
O [ 4.502 5.955 -0.000 ]
H [ 2.917 4.963 -0.000 ]
H [ 3.812 7.691 -0.000 ]
}
)
% basis set specification
basis<GaussianBasisSet>: (
name = "STO-3G"
molecule = $:molecule
)
mpqc: (
checkpoint = no
savestate = no
% method for computing the molecule's energy
mole<CLHF>: (
molecule = $:molecule
basis = $:basis
coor = $..:coor
memory = 16000000
)
% molecular coordinates for optimization
coor<SymmMolecularCoor>: (
molecule = $:molecule
generator<IntCoorGen>: (
molecule = $:molecule
extra_bonds = [ 2 5 ]
)
% use these instead of generated coordinates
variable<SetIntCoor>: [
<StreSimpleCo>:( atoms = [ 2 5 ] )
<BendSimpleCo>:( atoms = [ 2 5 4 ] )
<OutSimpleCo>: ( atoms = [ 5 2 1 3 ] )
<SumIntCoor>: (
coor: [
<StreSimpleCo>:( atoms = [ 1 2 ] )
<StreSimpleCo>:( atoms = [ 2 3 ] )
]
coef = [ 1.0 1.0 ]
)
<SumIntCoor>: (
coor: [
<StreSimpleCo>:( atoms = [ 4 5 ] )
<StreSimpleCo>:( atoms = [ 4 6 ] )
]
coef = [ 1.0 1.0 ]
)
<BendSimpleCo>:( atoms = [ 1 2 3 ] )
<BendSimpleCo>:( atoms = [ 5 4 6 ] )
]
% these are fixed by symmetry anyway,
fixed<SetIntCoor>: [
<SumIntCoor>: (
coor: [
<StreSimpleCo>:( atoms = [ 1 2 ] )
<StreSimpleCo>:( atoms = [ 2 3 ] )
]
coef = [ 1.0 -1.0 ]
)
<SumIntCoor>: (
coor: [
<StreSimpleCo>:( atoms = [ 4 5 ] )
<StreSimpleCo>:( atoms = [ 4 6 ] )
]
coef = [ 1.0 -1.0 ]
)
<TorsSimpleCo>:( atoms = [ 2 5 4 6] )
<OutSimpleCo>:( atoms = [ 3 2 6 4 ] )
<OutSimpleCo>:( atoms = [ 1 2 6 4 ] )
]
)
% optimizer object for the molecular geometry
opt<QNewtonOpt>: (
function = $..:mole
update<BFGSUpdate>: ()
convergence<MolEnergyConvergence>: (
cartesian = yes
energy = $..:..:mole
)
% give a partial guess hessian in internal coordinates
% the missing elements will be filled in automatically
hessian = [
[ 0.0109261670 ]
[ -0.0004214845 0.0102746106 ]
[ -0.0008600592 0.0030051330 0.0043149957 ]
[ 0.0 0.0 0.0 ]
[ 0.0 0.0 0.0 ]
[ 0.0 0.0 0.0 ]
[ 0.0 0.0 0.0 ]
]
)
)
Optimization with a Hydrogen Bond
The automatic internal coordinate generator will fail if it cannot find enough redundant internal coordinates. In this case, the internal coordinate generator must be explicitly created in the input and given extra connectivity information, as is shown below.
% emacs should use -*- KeyVal -*- mode
% molecule specification
molecule<Molecule>: (
symmetry = C1
{ atoms geometry } = {
H [ 0.088 2.006 1.438 ]
O [ 0.123 3.193 0.000 ]
H [ 0.088 2.006 -1.438 ]
O [ 4.502 5.955 -0.000 ]
H [ 2.917 4.963 -0.000 ]
H [ 3.812 7.691 -0.000 ]
}
)
% basis set specification
basis<GaussianBasisSet>: (
name = "STO-3G"
molecule = $:molecule
)
mpqc: (
checkpoint = no
savestate = no
% method for computing the molecule's energy
mole<CLHF>: (
molecule = $:molecule
basis = $:basis
coor = $..:coor
memory = 16000000
)
% molecular coordinates for optimization
coor<SymmMolecularCoor>: (
molecule = $:molecule
% give an internal coordinate generator that knows about the
% hydrogen bond between atoms 2 and 5
generator<IntCoorGen>: (
molecule = $:molecule
extra_bonds = [ 2 5 ]
)
)
% optimizer object for the molecular geometry
opt<QNewtonOpt>: (
function = $..:mole
update<BFGSUpdate>: ()
convergence<MolEnergyConvergence>: (
cartesian = yes
energy = $..:..:mole
)
)
)
Fixed Coordinate Optimization
This example shows how to selectively fix internal coordinates in an optimization. Any number of linearly independent coordinates can be given. These coordinates must remain linearly independent throughout the optimization, a condition that might not hold since the coordinates can be nonlinear.
By default, the initial fixed coordinates' values are taken from the cartesian geometry given by the Molecule object; however, the molecule will be displaced to the internal coordinate values given with the fixed internal coordinates if have_fixed_values keyword is set to true, as shown in this example. In this case, the initial cartesian geometry should be reasonably close to the desired initial geometry and all of the variable coordinates will be frozen to their original values during the initial displacement.
% emacs should use -*- KeyVal -*- mode
% molecule specification
molecule<Molecule>: (
symmetry = CS
{ atoms geometry } = {
H [ 3.04 -0.69 -1.59 ]
H [ 3.04 -0.69 1.59 ]
N [ 2.09 -0.48 -0.00 ]
C [ -0.58 -0.15 0.00 ]
H [ -1.17 1.82 0.00 ]
H [ -1.41 -1.04 -1.64 ]
H [ -1.41 -1.04 1.64 ]
}
)
% basis set specification
basis<GaussianBasisSet>: (
name = "3-21G*"
molecule = $:molecule
)
mpqc: (
checkpoint = no
savestate = no
% molecular coordinates for optimization
coor<SymmMolecularCoor>: (
molecule = $:molecule
generator<IntCoorGen>: (
molecule = $:molecule
)
have_fixed_values = yes
fixed<SetIntCoor>: [
<OutSimpleCo>: ( value = -0.1
label = "N-inversion"
atoms = [4 3 2 1] )
]
)
% method for computing the molecule's energy
mole<CLHF>: (
molecule = $:molecule
basis = $:basis
coor = $..:coor
memory = 16000000
)
% optimizer object for the molecular geometry
opt<QNewtonOpt>: (
max_iterations = 20
function = $..:mole
update<BFGSUpdate>: ()
convergence<MolEnergyConvergence>: (
cartesian = yes
energy = $..:..:mole
)
)
)
Transition State Optimization
This example shows a transition state optimization of the N-inversion in 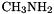 using mode following. The initial geometry was obtained by doing a few fixed coordinate optimizations along the inversion coordinate.
using mode following. The initial geometry was obtained by doing a few fixed coordinate optimizations along the inversion coordinate.
% emacs should use -*- KeyVal -*- mode
% molecule specification
molecule<Molecule>: (
symmetry = CS
{ atoms geometry } = {
H [ 3.045436 -0.697438 -1.596748 ]
H [ 3.045436 -0.697438 1.596748 ]
N [ 2.098157 -0.482779 -0.000000 ]
C [ -0.582616 -0.151798 0.000000 ]
H [ -1.171620 1.822306 0.000000 ]
H [ -1.417337 -1.042238 -1.647529 ]
H [ -1.417337 -1.042238 1.647529 ]
}
)
% basis set specification
basis<GaussianBasisSet>: (
name = "3-21G*"
molecule = $:molecule
)
mpqc: (
checkpoint = no
savestate = no
% molecular coordinates for optimization
coor<SymmMolecularCoor>: (
molecule = $:molecule
generator<IntCoorGen>: (
molecule = $:molecule
)
followed<OutSimpleCo> = [ "N-inversion" 4 3 2 1 ]
)
% method for computing the molecule's energy
mole<CLHF>: (
molecule = $:molecule
basis = $:basis
coor = $..:coor
memory = 16000000
)
% optimizer object for the molecular geometry
opt<EFCOpt>: (
transition_state = yes
mode_following = yes
max_iterations = 20
function = $..:mole
update<PowellUpdate>: ()
convergence<MolEnergyConvergence>: (
cartesian = yes
energy = $..:..:mole
)
)
)
Transition State Optimization with a Computed Guess Hessian
This example shows a transition state optimization of the N-inversion in using mode following. The initial geometry was obtained by doing a few fixed coordinate optimizations along the inversion coordinate. An approximate guess Hessian will be computed, which makes the optimiziation converge much faster in this case.
% emacs should use -*- KeyVal -*- mode
% molecule specification
molecule<Molecule>: (
symmetry = CS
{ atoms geometry } = {
H [ 3.045436 -0.697438 -1.596748 ]
H [ 3.045436 -0.697438 1.596748 ]
N [ 2.098157 -0.482779 -0.000000 ]
C [ -0.582616 -0.151798 0.000000 ]
H [ -1.171620 1.822306 0.000000 ]
H [ -1.417337 -1.042238 -1.647529 ]
H [ -1.417337 -1.042238 1.647529 ]
}
)
% basis set specification
basis<GaussianBasisSet>: (
name = "3-21G*"
molecule = $:molecule
)
mpqc: (
checkpoint = no
savestate = no
% molecular coordinates for optimization
coor<SymmMolecularCoor>: (
molecule = $:molecule
generator<IntCoorGen>: (
molecule = $:molecule
)
followed<OutSimpleCo> = [ "N-inversion" 4 3 2 1 ]
)
% method for computing the molecule's energy
mole<CLHF>: (
molecule = $:molecule
basis = $:basis
coor = $..:coor
memory = 16000000
guess_hessian<FinDispMolecularHessian>: (
molecule = $:molecule
only_totally_symmetric = yes
eliminate_cubic_terms = no
checkpoint = no
energy<CLHF>: (
molecule = $:molecule
memory = 16000000
basis<GaussianBasisSet>: (
name = "3-21G"
molecule = $:molecule
)
)
)
)
% optimizer object for the molecular geometry
opt<EFCOpt>: (
transition_state = yes
mode_following = yes
max_iterations = 20
function = $..:mole
update<PowellUpdate>: ()
convergence<MolEnergyConvergence>: (
cartesian = yes
energy = $..:..:mole
)
)
)
Hartree-Fock energy with intermediate checkpointing
The following two sections demonstrate how MPQC can be used to save the mole object periodically. This input will compute the Hartree-Fock energy of water while saving the mole object every 3 iterations.
% emacs should use -*- KeyVal -*- mode
% molecule specification
molecule<Molecule>: (
symmetry = C2V
unit = angstrom
{ atoms geometry } = {
O [ 0.00000000 0.00000000 0.37000000 ]
H [ 0.78000000 0.00000000 -0.18000000 ]
H [ -0.78000000 0.00000000 -0.18000000 ]
}
)
% basis set specification
basis<GaussianBasisSet>: (
name = "STO-3G"
molecule = $:molecule
)
mpqc: (
checkpoint = yes
filename = "h2o-rhf-STO3G"
checkpoint_freq = 3
savestate = no
% method for computing the molecule's energy
mole<CLHF>: (
molecule = $:molecule
basis = $:basis
memory = 16000000
)
)
The mole object will be saved to files named "h2o-rhf-STO3G.wfn.<iter#>.tmp" where <iter#> is the SCF iteration number (3, 6, etc.). Only the most recent file is kept, files from previous iterations are removed automatically. Keyword filename here is used to set the default file name prefix.
MP2-R12 energy with intermediate checkpointing
The following input will compute the MP2-R12 energy of water in standard approximation A' (MP2-R12/A') while saving the mole object at intermediate checkpoints.
% emacs should use -*- KeyVal -*- mode
% molecule specification
molecule<Molecule>: (
symmetry = C2V
unit = angstrom
{ atoms geometry } = {
O [ 0.00000000 0.00000000 0.37000000 ]
H [ 0.78000000 0.00000000 -0.18000000 ]
H [ -0.78000000 0.00000000 -0.18000000 ]
}
)
% basis set specification
basis<GaussianBasisSet>: (
name = "cc-pVDZ"
molecule = $:molecule
)
% auxiliary basis set specification
abasis<GaussianBasisSet>: (
name = "aug-cc-pVDZ"
molecule = $:molecule
)
mpqc: (
checkpoint = yes
filename = "h2o-mp2r12ap-vdz-avdz"
savestate = no
% method for computing the molecule's energy
mole<MBPT2_R12>: (
molecule = $:molecule
basis = $:basis
aux_basis = $:abasis
stdapprox = "A'"
nfzc = 1
memory = 16000000
integrals<IntegralCints>:()
% reference wavefunction
reference<CLHF>: (
molecule = $:molecule
basis = $:basis
memory = 16000000
integrals<IntegralCints>:()
)
)
)
The mole object will be saved to a file named h2o-mp2r12ap-vdz-avdz.wfn". Keyword filename here is used to set the default file name prefix. Objects of the MBPT2_R12 type are checkpointed after the HF procedure, after the first integrals (SBS) transformation, and after the optional second (ABS) transformation.
HF gradient computed from a previously computed HF wave funtion
The following will illustrate how to reuse previously computed MolecularEnergy objects in subsequent computations. The first input computes Hartree-Fock energy for water and saves the mole object to file h2o-rhf-sto3g.wfn.
% emacs should use -*- KeyVal -*- mode
% molecule specification
molecule<Molecule>: (
symmetry = C2V
unit = angstrom
{ atoms geometry } = {
O [ 0.00000000 0.00000000 0.37000000 ]
H [ 0.78000000 0.00000000 -0.18000000 ]
H [ -0.78000000 0.00000000 -0.18000000 ]
}
)
% basis set specification
basis<GaussianBasisSet>: (
name = "STO-3G"
molecule = $:molecule
)
mpqc: (
checkpoint = no
savestate = yes
filename = "h2o-rhf-sto3g"
% method for computing the molecule's energy
mole<CLHF>: (
molecule = $:molecule
basis = $:basis
memory = 16000000
)
)
The second input reuses the mole object from the previous run to compute the gradient of the Hartree-Fock energy.
% emacs should use -*- KeyVal -*- mode mpqc: ( checkpoint = no savestate = no restart = yes restart_file = "h2o-rhf-sto3g.wfn" do_gradient = yes )
MP2 Energy computed using precomputed Hartree-Fock wave function
The following input will compute the MP2 energy of water using a saved Hartree-Fock wave function obtained using the first input from HF gradient computed from a previously computed HF wave funtion.
% emacs should use -*- KeyVal -*- mode
% molecule specification
molecule<Molecule>: (
symmetry = C2V
unit = angstrom
{ atoms geometry } = {
O [ 0.00000000 0.00000000 0.37000000 ]
H [ 0.78000000 0.00000000 -0.18000000 ]
H [ -0.78000000 0.00000000 -0.18000000 ]
}
)
% basis set specification
basis<GaussianBasisSet>: (
name = "STO-3G"
molecule = $:molecule
)
% wave function file object specification
wfnfile<BcastStateInBin>:file = "h2o-rhf-sto3g.wfn"
mpqc: (
checkpoint = no
savestate = no
% method for computing the molecule's energy
mole<MBPT2>: (
molecule = $:molecule
basis = $:basis
memory = 16000000
% reference wavefunction
reference<SavableStateProxy>: (
statein = $:wfnfile
object = "CLHF"
)
)
)
Note that now object reference is of type SavableStateProxy, rather than CLHF. SavableStateProxy is a special object type that can be converted at runtime into the desired type (in this case, CLHF, as indicated by object).
CLHF energy using a CCA integrals component
The following input will compute the CLHF energy of water using a CCA integrals component via the IntegralCCA adaptor class.
% emacs should use -*- KeyVal -*- mode
% molecule specification
molecule<Molecule>: (
symmetry = C2V
unit = angstrom
{ atoms geometry } = {
O [ 0.00000000 0.00000000 0.37000000 ]
H [ 0.78000000 0.00000000 -0.18000000 ]
H [ -0.78000000 0.00000000 -0.18000000 ]
}
)
% basis set specification
basis<GaussianBasisSet>: (
name = "STO-3G"
molecule = $:molecule
)
mpqc: (
% path to component libraries
cca_path = /usr/local/lib/cca
% sidl class names of components which will be instantiated
cca_load = MPQC.IntegralEvaluatorFactory
do_cca = yes
checkpoint = no
savestate = no
% method for computing the molecule's energy
mole<CLHF>: (
molecule = $:molecule
basis = $:basis
% cca integrals adaptor class
integrals<IntegralCCA>: (
molecule = $:molecule
% integral buffer type
integral_buffer = opaque
% integral package
integral_package = intv3
% factory component sidl class name
evaluator_factory = MPQC.IntegralEvaluatorFactory
)
)
)