Computes the local second order perturbation theory energy. More...
#include <chemistry/qc/lmp2/lmp2.h>
Public Member Functions | |
| LMP2 (const sc::Ref< sc::KeyVal > &) | |
| Construct an LMP2 object from KeyVal input. More... | |
| LMP2 (sc::StateIn &) | |
| void | save_data_state (sc::StateOut &) |
| Save the base classes (with save_data_state) and the members in the same order that the StateIn CTOR initializes them. More... | |
| void | compute (void) |
| Recompute at least the results that have compute true and are not already computed. More... | |
| int | nelectron (void) |
| Returns the number of electrons. | |
| sc::RefSymmSCMatrix | density (void) |
| Returns the SO density. | |
| double | magnetic_moment () const |
Computes the S (or J) magnetic moment of the target state(s), in units of 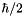 . More... . More... | |
| int | value_implemented (void) const |
 Public Member Functions inherited from sc::LCorr Public Member Functions inherited from sc::LCorr | |
| LCorr (const sc::Ref< sc::KeyVal > &) | |
| Construct an LCorr object from KeyVal input. More... | |
| LCorr (sc::StateIn &) | |
| Public Member Functions inherited from sc::Wavefunction | |
| Wavefunction (StateIn &) | |
| Wavefunction (const Ref< KeyVal > &) | |
| The KeyVal constructor. More... | |
| void | save_data_state (StateOut &) |
| Save the base classes (with save_data_state) and the members in the same order that the StateIn CTOR initializes them. More... | |
| double | density (const SCVector3 &) |
| double | density_gradient (const SCVector3 &, double *) |
| double | natural_orbital (const SCVector3 &r, int iorb) |
| double | natural_orbital_density (const SCVector3 &r, int orb, double *orbval=0) |
| double | orbital (const SCVector3 &r, int iorb, const RefSCMatrix &orbs) |
| void | orbitals (const SCVector3 &r, const RefSCMatrix &orbs, RefSCVector &values) |
| double | orbital_density (const SCVector3 &r, int iorb, const RefSCMatrix &orbs, double *orbval=0) |
| double | total_charge () const |
| Returns the total charge of the system. | |
| virtual RefSymmSCMatrix | ao_density () |
| Returns the AO density. | |
| virtual RefSCMatrix | natural_orbitals () |
| Returns the natural orbitals, in SO basis. | |
| virtual RefDiagSCMatrix | natural_density () |
| Returns the natural density (a diagonal matrix). | |
| int | spin_polarized () |
| Return 1 if the magnetic moment != 0. | |
| int | dk () const |
| Returns the level the of the Douglas-Kroll approximation. | |
| virtual RefSymmSCMatrix | alpha_density () |
| Return alpha electron densities in the SO basis. | |
| virtual RefSymmSCMatrix | beta_density () |
| Return beta electron densities in the SO basis. | |
| virtual RefSymmSCMatrix | alpha_ao_density () |
| Return alpha electron densities in the AO basis. | |
| virtual RefSymmSCMatrix | beta_ao_density () |
| Return beta electron densities in the AO basis. | |
| virtual RefSCMatrix | nao (double *atom_charges=0) |
| returns the ao to nao transformation matrix | |
| virtual RefSymmSCMatrix | overlap () |
| Returns the SO overlap matrix. | |
| virtual RefSymmSCMatrix | core_hamiltonian_for_basis (const Ref< GaussianBasisSet > &bas, const Ref< GaussianBasisSet > &pbas=0) |
| Returns the SO core Hamiltonian in the given basis and momentum basis. More... | |
| virtual RefSymmSCMatrix | core_hamiltonian () |
| Returns the SO core Hamiltonian. | |
| RefSymmSCMatrix | core_hamiltonian_nr (const Ref< GaussianBasisSet > &bas) |
| virtual double | nuclear_repulsion_energy () |
| Returns the nuclear repulsion energy. More... | |
| void | nuclear_repulsion_energy_gradient (double *g) |
| Computes the nuclear repulsion gradient. More... | |
| virtual void | nuclear_repulsion_energy_gradient (double **g) |
| Computes the nuclear repulsion gradient. More... | |
| RefSCDimension | ao_dimension () |
| Atomic orbital dimension. | |
| RefSCDimension | so_dimension () |
| Symmetry adapted orbital dimension. | |
| RefSCDimension | oso_dimension () |
| Orthogonalized symmetry adapted orbital dimension. | |
| Ref< SCMatrixKit > | basis_matrixkit () |
| Matrix kit for AO, SO, orthogonalized SO, and MO dimensioned matrices. | |
| Ref< Molecule > | molecule () const |
| Returns the Molecule. | |
| Ref< GaussianBasisSet > | basis () const |
| Returns the basis set. | |
| Ref< GaussianBasisSet > | momentum_basis () const |
| Returns the basis used for p^2 in the DK correction. | |
| Ref< GaussianBasisSet > | atom_basis () const |
| Returns the basis set describing the nuclear charge distributions. | |
| const double * | atom_basis_coef () const |
| Returns the coefficients of the nuclear charge distribution basis functions. | |
| Ref< Integral > | integral () |
| Returns the integral evaluator. | |
| void | symmetry_changed () |
| Call this if you have changed the molecular symmetry of the molecule contained by this MolecularEnergy. | |
| RefSCMatrix | so_to_orthog_so () |
| Returns a matrix which does the default transform from SO's to orthogonal SO's. More... | |
| RefSCMatrix | so_to_orthog_so_inverse () |
| Returns the inverse of the transformation returned by so_to_orthog_so. | |
| OverlapOrthog::OrthogMethod | orthog_method () const |
| Returns the orthogonalization method. | |
| virtual void | set_orthog_method (const OverlapOrthog::OrthogMethod &) |
| (Re)Sets the orthogonalization method and makes this obsolete. More... | |
| double | lindep_tol () const |
| Returns the tolerance for linear dependencies. | |
| void | set_lindep_tol (double) |
| Re(Sets) the tolerance for linear dependencies. | |
| void | obsolete () |
| Marks all results as being out of date. More... | |
| void | print (std::ostream &=ExEnv::out0()) const |
| Print information about the object. | |
| void | writeorbitals () |
| output orbitals to some files to facilitate plotting, with the help of the WriteOrbital class. | |
| Public Member Functions inherited from sc::MolecularEnergy | |
| MolecularEnergy (const MolecularEnergy &) | |
| MolecularEnergy (const Ref< KeyVal > &) | |
| The KeyVal constructor. More... | |
| MolecularEnergy (StateIn &) | |
| void | set_checkpoint () |
| Set up checkpointing. | |
| void | set_checkpoint_file (const char *) |
| void | set_checkpoint_freq (int freq) |
| bool | if_to_checkpoint () const |
| Check if need to checkpoint. | |
| const char * | checkpoint_file () const |
| int | checkpoint_freq () const |
| MolecularEnergy & | operator= (const MolecularEnergy &) |
| virtual double | energy () |
| A wrapper around value();. | |
| virtual RefSCDimension | moldim () const |
| void | guess_hessian (RefSymmSCMatrix &) |
| Compute a quick, approximate hessian. | |
| RefSymmSCMatrix | inverse_hessian (RefSymmSCMatrix &) |
| int | gradient_implemented () const |
| Reports whether gradient is implemented either analytically or using MolecularGradient object. More... | |
| int | hessian_implemented () const |
| Reports whether hessian is implemented either analytically or using MolecularHessian object. More... | |
| void | set_desired_gradient_accuracy (double acc) |
| These functions overload their Function counterparts. More... | |
| void | set_desired_hessian_accuracy (double acc) |
| void | set_molhess (const Ref< MolecularHessian > &molhess) |
| Use this function to provide MolecularHessian object that will be used to compute hessian. More... | |
| const Ref< MolecularHessian > & | molhess () const |
| RefSymmSCMatrix | hessian () |
| Will throw if hessian_implemented() returns 0. | |
| void | set_molgrad (const Ref< MolecularGradient > &molgrad) |
| Use this function to provide MolecularGradient object that will be used to compute gradient. More... | |
| const Ref< MolecularGradient > & | molgrad () const |
| RefSCVector | gradient () |
| Will throw if gradient_implemented() returns 0. | |
| void | set_x (const RefSCVector &) |
| Set and retrieve the coordinate values. | |
| RefSCVector | get_cartesian_x () |
| Return the cartesian coordinates. | |
| RefSCVector | get_cartesian_gradient () |
| Return the cartesian gradient. | |
| RefSymmSCMatrix | get_cartesian_hessian () |
| Return the cartesian hessian. | |
| Ref< MolecularCoor > | molecularcoor () |
| Ref< NonlinearTransform > | change_coordinates () |
| An optimizer can call change coordinates periodically to give the function an opportunity to change its coordinate system. More... | |
| virtual void | purge () |
| This function purges any caches of data in MolecularEnergy. More... | |
| const RefSCVector & | electric_field () const |
| returns the electric field vector | |
| void | print_natom_3 (const RefSCVector &, const char *t=0, std::ostream &o=ExEnv::out0()) const |
| Nicely print n x 3 data that are stored in a vector. | |
| void | print_natom_3 (double **, const char *t=0, std::ostream &o=ExEnv::out0()) const |
| void | print_natom_3 (double *, const char *t=0, std::ostream &o=ExEnv::out0()) const |
| Public Member Functions inherited from sc::Function | |
| int | gradient_needed () const |
| int | do_gradient (int) |
| virtual double | actual_gradient_accuracy () const |
| virtual double | desired_gradient_accuracy () const |
| AccResultRefSCVector & | gradient_result () |
| int | hessian_needed () const |
| int | do_hessian (int) |
| virtual double | actual_hessian_accuracy () const |
| virtual double | desired_hessian_accuracy () const |
| AccResultRefSymmSCMatrix & | hessian_result () |
| virtual bool | desired_value_accuracy_set_to_default () const |
| virtual bool | desired_gradient_accuracy_set_to_default () const |
| virtual bool | desired_hessian_accuracy_set_to_default () const |
| RefSCVector | get_x () const |
| const RefSCVector & | get_x_no_copy () const |
| void | print_desired_accuracy (std::ostream &=ExEnv::out0()) const |
| similar to print(), but only prins desired accuracies | |
| virtual bool | throw_if_tolerance_exceeded () const |
| Overridden Compute member. | |
| Function () | |
| Function (StateIn &) | |
| Function (const Function &) | |
| Function (const Ref< KeyVal > &, double funcacc=DBL_EPSILON, double gradacc=DBL_EPSILON, double hessacc=DBL_EPSILON) | |
| The keyval constructor reads the following keywords: More... | |
| virtual | ~Function () |
| Function & | operator= (const Function &) |
| Ref< SCMatrixKit > | matrixkit () const |
| Return the SCMatrixKit used to construct vectors and matrices. | |
| RefSCDimension | dimension () const |
| Return the SCDimension of the problem. | |
| virtual double | value () |
| Return the value of the function. | |
| int | value_needed () const |
| Returns nonzero if the current value is not up-to-date. | |
| int | do_value (int) |
| If passed a nonzero number, compute the value the next time compute() is called. More... | |
| AccResultdouble & | value_result () |
| virtual void | set_desired_value_accuracy (double) |
| Set the accuracy to which the value is to be computed. | |
| virtual double | actual_value_accuracy () const |
| Return the accuracy with which the value has been computed. | |
| virtual double | desired_value_accuracy () const |
| Return the accuracy with which the value is to be computed. | |
| Public Member Functions inherited from sc::SavableState | |
| SavableState & | operator= (const SavableState &) |
| void | save_state (StateOut &) |
| Save the state of the object as specified by the StateOut object. More... | |
| void | save_object_state (StateOut &) |
| This can be used for saving state when the exact type of the object is known for both the save and the restore. More... | |
| virtual void | save_vbase_state (StateOut &) |
| Save the virtual bases for the object. More... | |
| Public Member Functions inherited from sc::DescribedClass | |
| DescribedClass (const DescribedClass &) | |
| DescribedClass & | operator= (const DescribedClass &) |
| ClassDesc * | class_desc () const MPQC__NOEXCEPT |
| This returns the unique pointer to the ClassDesc corresponding to the given type_info object. More... | |
| const char * | class_name () const |
| Return the name of the object's exact type. | |
| int | class_version () const |
| Return the version of the class. | |
| Ref< DescribedClass > | ref () |
| Return this object wrapped up in a Ref smart pointer. More... | |
| Public Member Functions inherited from sc::RefCount | |
| size_t | identifier () const |
| Return the unique identifier for this object that can be compared for different objects of different types. More... | |
| int | lock_ptr () const |
| Lock this object. | |
| int | unlock_ptr () const |
| Unlock this object. | |
| void | use_locks (bool inVal) |
| start and stop using locks on this object | |
| refcount_t | nreference () const |
| Return the reference count. | |
| refcount_t | reference () |
| Increment the reference count and return the new count. | |
| refcount_t | dereference () |
| Decrement the reference count and return the new count. | |
| int | managed () const |
| void | unmanage () |
| Turn off the reference counting mechanism for this object. More... | |
Additional Inherited Members | |
| Static Public Member Functions inherited from sc::Wavefunction | |
| static void | orbitals (const Ref< OrbitalSpace > &orbs, const std::vector< SCVector3 > &r, std::vector< double > &values) |
| Static Public Member Functions inherited from sc::SavableState | |
| static void | save_state (SavableState *s, StateOut &) |
| static SavableState * | restore_state (StateIn &si) |
| Restores objects saved with save_state. More... | |
| static SavableState * | key_restore_state (StateIn &si, const char *keyword) |
| Like restore_state, but keyword is used to override values while restoring. | |
| static SavableState * | dir_restore_state (StateIn &si, const char *objectname, const char *keyword=0) |
| Protected Member Functions inherited from sc::LCorr | |
| int | n_unique_W () const |
| sma2::Array< 2 > & | unique_W (const domainmapvirbs_t &i) |
| std::vector< double > & | unique_eigvals (const domainmapvirbs_t &i) |
| sma2::Array< 2 > & | unique_F_tilde (const domainmapvirbs_t &i) |
| void | init_virb_to_bfns (const sma2::Range &vir) |
| This must be called before compute_W is called. | |
| void | compute_W (domainmapvirbs_t &virset, const sc::Ref< sc::GaussianBasisSet > &basis, sc::RefSCMatrix &F_vir_mat, sc::RefSCMatrix &S_mat, const sma2::Range &vir, int nocc_act, double bound) |
| void | transform_array (sma2::Array< 2 > &A, sma2::Array< 2 > &B, sma2::Array< 2 > &C, sma2::Array< 2 > &D, const sc::Ref< sc::MessageGrp > &msg) |
| Transform A to D using transformation matrices B,C: D = B^T*A*C. | |
| void | clear () |
| Release stored data. | |
| void | print_parameters () const |
| Print input parameters. | |
| Protected Member Functions inherited from sc::Wavefunction | |
| double | min_orthog_res () |
| double | max_orthog_res () |
| void | copy_orthog_info (const Ref< Wavefunction > &) |
| Protected Member Functions inherited from sc::MolecularEnergy | |
| void | failure (const char *) |
| virtual void | set_energy (double) |
| This is just a wrapper around set_value(). | |
| virtual void | set_gradient (RefSCVector &) |
| These are passed gradients and hessian in cartesian coordinates. More... | |
| virtual void | set_hessian (RefSymmSCMatrix &) |
| void | x_to_molecule () |
| void | molecule_to_x () |
| virtual bool | analytic_gradient_implemented () const |
| must overload this in a derived class if analytic gradient can be computed More... | |
| virtual bool | analytic_hessian_implemented () const |
| must overload this in a derived class if analytic hessian can be computed More... | |
| Protected Member Functions inherited from sc::Function | |
| virtual void | set_value (double) |
| virtual void | set_matrixkit (const Ref< SCMatrixKit > &) |
| Set the SCMatrixKit that should be used to construct the requisite vectors and matrices. | |
| virtual void | set_dimension (const RefSCDimension &) |
| virtual void | set_actual_value_accuracy (double) |
| virtual void | set_actual_gradient_accuracy (double) |
| virtual void | set_actual_hessian_accuracy (double) |
| RefSCVector & | get_x_reference () |
| Get read/write access to the coordinates for modification. | |
| void | do_change_coordinates (const Ref< NonlinearTransform > &) |
| Change the coordinate system and apply the given transform to intermediates matrices and vectors. | |
| Protected Member Functions inherited from sc::SavableState | |
| SavableState (const SavableState &) | |
| SavableState (StateIn &) | |
| Each derived class StateIn CTOR handles the restore corresponding to calling save_object_state, save_vbase_state, and save_data_state listed above. More... | |
| Protected Member Functions inherited from sc::RefCount | |
| RefCount (const RefCount &) | |
| RefCount & | operator= (const RefCount &) |
| Protected Attributes inherited from sc::Wavefunction | |
| ResultRefSCMatrix | natural_orbitals_ |
| ResultRefDiagSCMatrix | natural_density_ |
| Ref< GaussianBasisSet > | gbs_ |
| int | debug_ |
| Protected Attributes inherited from sc::MolecularEnergy | |
| Ref< PointGroup > | initial_pg_ |
| int | print_molecule_when_changed_ |
| Protected Attributes inherited from sc::Function | |
| Ref< SCMatrixKit > | matrixkit_ |
| Used to construct new matrices. | |
| RefSCVector | x_ |
| The variables. | |
| RefSCDimension | dim_ |
| The dimension of x_. | |
| AccResultdouble | value_ |
| The value of the function at x_. | |
| AccResultRefSCVector | gradient_ |
| The gradient at x_. | |
| AccResultRefSymmSCMatrix | hessian_ |
| The hessian at x_. | |
| bool | desired_value_accuracy_set_to_default_ |
| bool | desired_gradient_accuracy_set_to_default_ |
| bool | desired_hessian_accuracy_set_to_default_ |
| bool | throw_if_tolerance_exceeded_ |
Detailed Description
Computes the local second order perturbation theory energy.
This class implements a massively parallel algorithm for the quantum mechanical computation of energies of molecular systems using local second-order Møller-Plesset (LMP2) perturbation theory. It is built upon the Massively Parallel Quantum Chemistry program (MPQC). Both the storage requirement and the computational time scale linearly with the molecular size. High parallel efficiency of the algorithm has been demonstrated for applications employing up to 100 processors.
The parallel algorithm is designed to be scalable, employing a distributed data scheme for the two-electron integrals, avoiding communication bottlenecks, and distributing tasks in all computationally significant steps. A sparse data representation and a set of generalized contraction routines have been developed to allow efficient massively parallel implementation using distributed sparse multidimensional arrays.
The implementation of this software and its performance are described in Nielsen, I. M. B.; Janssen, C. L.; J. Chem. Theory and Comput.; 2007; pp. 71-79.
Constructor & Destructor Documentation
◆ LMP2()
| sc::LMP2::LMP2 | ( | const sc::Ref< sc::KeyVal > & | ) |
Construct an LMP2 object from KeyVal input.
This reads the keywords in the table below. The LCorr input has additional keywords that are also read.
| Keyword | Type | Default | Description |
reference | object | none | Gives an object that specializes OneBodyWavefunction which can be used as a reference LMP2 wavefunction. This keyword is required and has no default. |
max_iter | int | 100 | The maximum number of iterations that will be used to converge the LMP2 amplitudes. |
nfzc | int or auto | 0 | The number of frozen core orbitals. An integer can be specified or |
| | |
|
| | |
|
In addition to the above options, are a variety of options that is useful to developers, listed in the following table.
| Keyword | Type | Default | Description |
S_threshold | double | 1.0e-6 | The threshold used to determine which blocks of the overlap matrix are stored. |
integral_threshold | double | 1.0e-8 | A threshold used to prune the number of two electron integrals which are computed. |
q1_threshold | double | 1.0e-8 | A threshold used to prune the number of first quarter transformed integrals which are computed. |
q2_threshold | double | 1.0e-8 | A threshold used to prune the number of second quarter transformed integrals which are computed. |
q3_threshold | double | 1.0e-8 | A threshold used to prune the number of third quarter transformed integrals which are computed. |
q4_threshold | double | 1.0e-8 | A threshold used to prune the number of fourth quarter transformed integrals which are computed. |
threshold_factor | double | 10.0 | The desired energy tolerance is divided by this factor to obtain the threshold that determines which blocks of a variety of arrays are stored. |
complete_domains | bool | 0 | If true, all possible blocks are computed. The result in this case should be the same as the canonical MP2 energy. |
single_virtual_block | bool | 0 | All of the projected atomic virtual orbitals are grouped into a single, large block, rather than atom based blocks. |
i_ge_j | bool | 1 | If true, use i >= j in the formation of K_2occ. |
m_ge_n | bool | 1 | If true, use m >= n in the formation of K_2occ. |
minimize_q2 | bool | 1 | If this and |
always_use_dist_t | bool | 0 | If true, distribute the amplitudes, in which case an extra communication step is required each iteration. |
extrap_T | object | DIIS | This gives an object derived from SelfConsistentExtrapolation that will be used to extraplote the amplitudes. By default DIIS(6,8,0.0,2,1) will be used. |
occ_orbitals | string | pipek-mezey | The method used to provide the occupied orbitals. This can be |
vir_orbitals | string | projected_atomic | The method used to provide the virtual orbitals. This can be |
Member Function Documentation
◆ compute()
|
virtual |
Recompute at least the results that have compute true and are not already computed.
This should only be called by Result's members.
Implements sc::Compute.
◆ magnetic_moment()
|
virtual |
Computes the S (or J) magnetic moment of the target state(s), in units of .
Can be evaluated from density and overlap, as;
but derived Wavefunction may have this value as user input.
- Returns
- the magnetic moment
Reimplemented from sc::Wavefunction.
◆ save_data_state()
|
virtual |
◆ value_implemented()
|
virtual |
- Returns
- 1 (value implemented) or 0 (value not implemented, default). Must be overridden for the class to be useful.
Reimplemented from sc::Function.
The documentation for this class was generated from the following file:
- src/lib/chemistry/qc/lmp2/lmp2.h